1. Nativi-Nicolau JN, Karam C, Khella S, et al. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2022;27(3):785-793. https://doi.org/10.1007/s10741-021-10080-2
2. Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020;21(1):198. https://doi.org/10.1186/s12875-020-01252-4
3. Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709-716. https://doi.org/10.1016/j.jchf.2019.04.010
4. Bentellis I, Amarenco G, Gamé X, et al. Diagnosis and treatment of urinary and sexual dysfunction in hereditary TTR amyloidosis. Clin Auton Res. 2019;29(suppl 1):65-74. https://doi.org/10.1007/s10286-019-00627-7
5. Martins AC, Rosa AM, Costa E, et al. Ocular manifestations and therapeutic options in patients with familial amyloid polyneuropathy: a systematic review. Biomed Res Int. 2015;2015:282405. https://doi.org/10.1155/2015/282405
6. Kittleson MM, Ruberg FL, Ambardekar AV, et al; Writing Committee. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2023;81(11):1076-1126. https://doi.org/10.1016/j.jacc.2022.11.022
7. Kapoor M, Rossor AM, Laura M, et al. Clinical presentation, diagnosis and treatment of TTR amyloidosis. J Neuromuscul Dis. 2019;6(2):189-199. https://doi.org/10.3233/JND-180371
8. Garcia-Pavia P, Bengel F, Brito D, et al. Expert consensus on the monitoring of transthyretin amyloid cardiomyopathy. Eur J Heart Fail. 2021;23(6):895-905. https://doi.org/10.1002/ejhf.2198
9. Palma JA, Gonzalez-Duarte A, Kaufmann H. Orthostatic hypotension in hereditary transthyretin amyloidosis: epidemiology, diagnosis and management. Clin Auton Res. 2019;29(suppl 1):33-44. https://doi.org/10.1007/s10286-019-00623-x
10. Loavenbruck AJ, Singer W, Mauermann ML, et al. Transthyretin amyloid neuropathy has earlier neural involvement but better prognosis than primary amyloid counterpart: an answer to the paradox? Ann Neurol. 2016;80(3):401-411. https://doi.org/10.1002/ana.24725
11. Planté-Bordeneuve V, Ferreira A, Lalu T, et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology. 2007;69(7):693-698. https://doi.org/10.1212/01.wnl.0000267338.45673.f4
12. Gendre T, Planté-Bordeneuve V. Strategies to improve the quality of life in patients with hereditary transthyretin amyloidosis (hATTR) and autonomic neuropathy. Clin Auton Res. 2019;29(suppl 1):25-31. https://doi.org/10.1007/s10286-019-00624-w
13. Dispenzieri A, Coelho T, Conceição I, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022;17(1):236. https://doi.org/10.1186/s13023-022-02359-w
14. Lousada I, Maurer MS, Warner M, et al. Amyloidosis Research Consortium cardiac amyloidosis survey: results from patients with AL and ATTR amyloidosis and their caregivers [poster]. Presented at: XVI International Symposium on Amyloidosis (ISA); March 26-29, 2018; Kumamoto, Japan.
15. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. https://doi.org/10.1186/1750-1172-8-31
16. aus dem Siepen F, Hein S, Prestel S, et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019;108(12):1324-1330. https://doi.org/10.1007/s00392-019-01467-1
17. Karam C, Dimitrova D, Christ M, et al. Carpal tunnel syndrome and associated symptoms as first manifestation of hATTR amyloidosis. Neurol Clin Pract. 2019;9(4):309-313. https://doi.org/10.1212/CPJ.0000000000000640
18. Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872-2891. https://doi.org/10.1016/j.jacc.2019.04.003
19. Aldinc E, Campbell C, Gustafsson F, et al. Musculoskeletal manifestations associated with transthyretin-mediated (ATTR) amyloidosis: a systematic review. BMC Musculoskelet Disord. 2023;24(1):751. https://doi.org/10.1186/s12891-023-06853-5
20. Westermark P, Westermark GT, Suhr OB, et al. Transthyretin-derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci. 2014;119(3):223-228. https://doi.org/10.3109/03009734.2014.895786
21. Rubin J, Alvarez J, Teruya S, et al. Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: can we identify affected patients earlier? Amyloid. 2017;24(4):226-230. https://doi.org/10.1080/13506129.2017.1375908
22. Cortese A, Vegezzi E, Lozza A, et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017;88(5):457-458. https://doi.org/10.1136/jnnp-2016-315262
23. Lin X, Yarlas A, Vera-Llonch M, et al. Rate of neuropathic progression in hereditary transthyretin amyloidosis with polyneuropathy and other peripheral neuropathies: a systematic review and meta-analysis. BMC Neurol. 2021;21(1):70. https://doi.org/10.1186/s12883-021-02094-y
24. Karam C, Mauermann ML, Gonzalez-Duarte A, et al. Diagnosis and treatment of hereditary transthyretin amyloidosis with polyneuropathy in the United States: recommendations from a panel of experts. Muscle Nerve. 2024;69(3):273-287. https://doi.org/10.1002/mus.28026
25. Conceição I, González-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5-9. https://doi.org/10.1111/jns.12153
26. Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109-2122. https://doi.org/10.1007/s00415-019-09688-0
27. Gonzalez-Duarte A, Valdés-Ferrer SI, Cantú-Brito C. Characteristics and natural history of autonomic involvement in hereditary ATTR amyloidosis: a systematic review. Clin Auton Res. 2019;29(suppl 1):1-9. https://doi.org/10.1007/s10286-019-00630-y
28. Sekijima Y, Ueda M, Koike H, et al. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6. https://doi.org/10.1186/s13023-017-0726-x
29. Barroso FA, Coelho T, Dispenzieri A, et al. Characteristics of patients with autonomic dysfunction in the Transthyretin Amyloidosis Outcomes Survey (THAOS). Amyloid. 2022;29(3):175-183. https://doi.org/10.1080/13506129.2022.2043270
30. Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12(9):e006075. https://doi.org/10.1161/CIRCHEARTFAILURE.119.006075
31. Hahn VS, Yanek LR, Vaishnav J, et al. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail. 2020;8(9):712-724. https://doi.org/10.1016/j.jchf.2020.04.007
32. Ruberg FL, Maurer MS. Cardiac amyloidosis due to transthyretin protein: a review. JAMA. 2024;331(9):778-791. https://doi.org/10.1001/jama.2024.0442
33. Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222-228.e1. https://doi.org/10.1016/j.ahj.2012.04.015
34. Sabbour H, Hasan KY, Al Badarin F, et al. From clinical clues to final diagnosis: the return of detective work to clinical medicine in cardiac amyloidosis. Front Cardiovasc Med. 2021;8:644508. https://doi.org/10.3389/fcvm.2021.644508
35. Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol. 2017;70(4):466-477. https://doi.org/10.1016/j.jacc.2017.05.053
36. Bazell C, Alston M, Kumar N, et al. Descriptive characteristics of patients diagnosed with transthyretin amyloidosis (ATTR) in the Medicare fee-for-service and commercial populations [poster]. Presented at: International Symposium of Amyloidosis (ISA); May 26-30, 2024; Rochester, MN. Poster 518. https://doi.org/10.26226/m.65f9bf8be6f73964e1d4f804
37. Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-172. https://doi.org/10.1016/j.jacc.2016.03.596
38. Wixner J, Mundayat R, Karayal ON, et al. THAOS: gastrointestinal manifestations of transthyretin amyloidosis – common complications of a rare disease. Orphanet J Rare Dis. 2014;9:61. https://doi.org/10.1186/1750-1172-9-61
39. Chandrashekar P, Alhuneafat L, Mannello M, et al. Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: a systematic review. Circ Genom Precis Med. 2021;14(5):e003356. https://doi.org/10.1161/CIRCGEN.121.003356







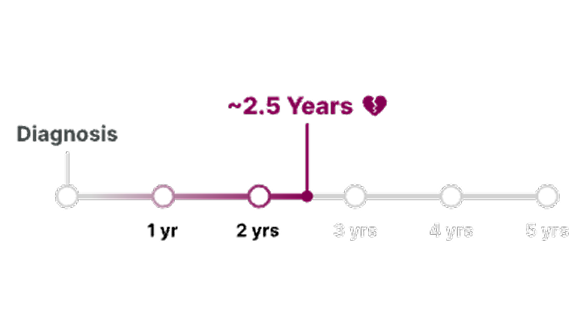














































 What is an AstraZeneca authorized person?
What is an AstraZeneca authorized person?